
Machine Learning Is Transforming Analytical Chemistry
Machine learning is revolutionising our strategies to tackle complex problems in many fields: From predicting economic trends to diagnosing diseases, machine learning has become a pivotal tool. In analytical chemistry, machine learning is increasingly being used to predict key properties of small molecules, which is crucial for drug development and other applications. Such predictions include determining if a new drug could be toxic before testing it on animals or humans, assessing how well a molecule binds to another molecule, or understanding how a drug is absorbed, distributed, metabolized, and excreted within the body.
Training Data Limits Are Model Limits
However, like any powerful tool, machine learning comes with its challenges. To avoid misapplying methods or misinterpreting results, machine learning requires substantial background knowledge and adherence to rigorous guidelines to ensure its reliability in chemistry and life sciences. One hidden risk is poorly prepared training data. Just as a chef can’t create a delicious meal with subpar ingredients, a machine learning model can’t make accurate predictions with biased or unrepresentative data. The issue of coverage bias in machine learning is a growing concern. This bias can significantly limit a model’s predictive power and lead to misleading results. Training a model on a specific subset of molecules limits its generalisability to that subset. In other words, if a model is trained solely on, say, lipids, it’s likely to perform poorly on the broader chemical space.
End-to-End Machine Learning Models: Power or Pitfall?
A rising trend in machine learning is the development of end-to-end models that avoid explicit domain knowledge integration1. These models try to learn everything from the data itself without needing extra information. While powerful, they operate under the assumption of unbiased training and evaluation data. When applied to biased datasets, end-to-end models may exhibit exceptional performance within the confines of that data but struggle to generalise to unseen data. Without careful consideration of the domain of applicability, the impressive performance of end-to-end models on biased data can create a false sense of confidence, potentially leading to inaccurate predictions and flawed conclusions.
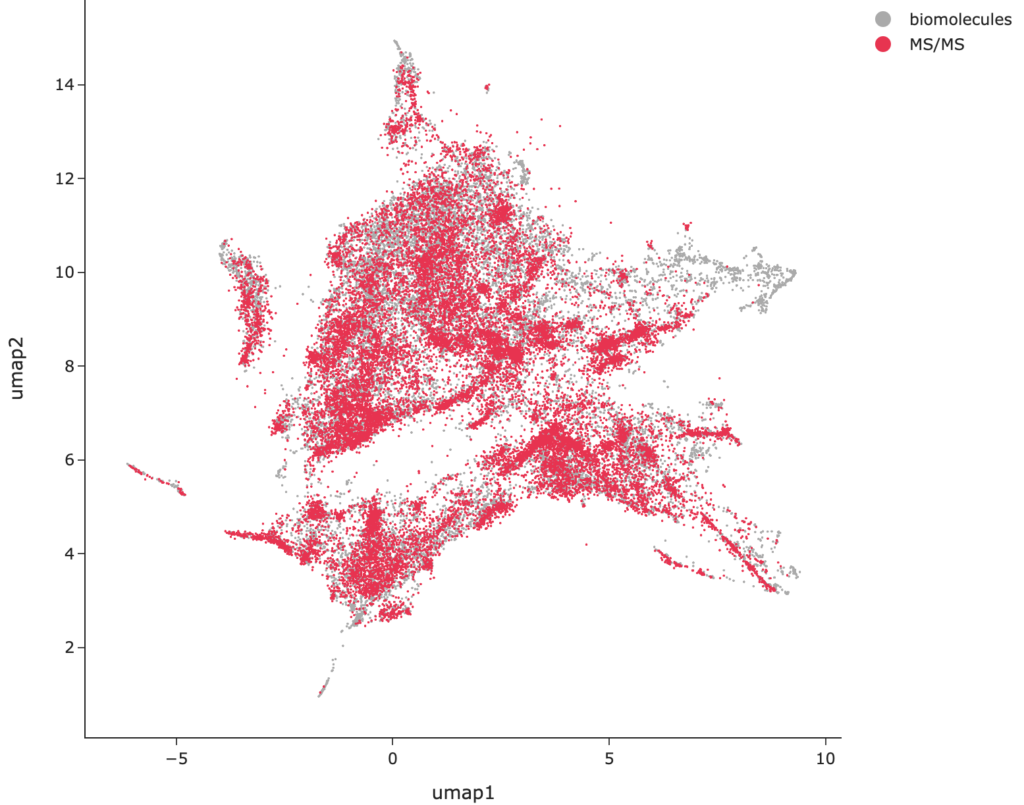
Measuring Training Data Distribution By Visualizing The Molecular Space
In small molecule machine learning, the training datasets are often influenced by factors like compound availability and cost. To evaluate the quality of a dataset, researchers need to check whether the dataset is a uniform subsample of the biomolecular space. One possibility is to use dimensionality reduction techniques to project the data into 2-dimensional space and visually inspect whether the training data covers the same regions as the full dataset. UMAP (Uniform Manifold Approximation and Projection) embeddings2 visualize complex data by arranging it in a way that makes it easier to see patterns and relationships among them. They can be used to visualize the universe of biomolecular structures in a 2-dimensional plot. This requires a metric of similarity or dissimilarity between molecular structures. The Maximum Common Edge Subgraph distance is the minimum number of bonds (edges) we have to remove from two molecular structures (graphs) so that the resulting structures are isomorphic. We can think of it as the number of chemical reactions required to transform one molecule into another. Finding the Maximum Common Edge Subgraph (MCES) is a computationally hard problem. To compute distances for the UMAP embedding requires to swiftly solve millions of instances of this problem. The groups of S. Böcker and G. W. Klau have developed an efficient approach combining Integer Linear Programming and heuristic bounds to solve this problem3.
Why Not Use Tanimoto Coefficients?
The Tanimoto coefficient4 is a widely used method for measuring the similarity between molecular structures. It is computationally efficient and facilitates the swift processing of large datasets. However, this method requires transforming molecular structures into binary vectors, known as molecular fingerprints, which inevitably leads to a loss of substantial information. Critiques of the Tanimoto coefficient5 date back to 1998, highlighting its limitations. One significant issue is that the similarity values it generates often deviate from chemical intuition. For instance, two molecules with high perceived structural similarity may yield a low Tanimoto coefficient, while dissimilar molecules might show unexpectedly high similarity scores. These inconsistencies are independent from the molecular fingerprint used and similar issues occur also for other coefficients that rely on binary vector encoding. Thus, while Tanimoto coefficients are convenient, they may not always provide an accurate representation of molecular similarity. The MCES better captures the chemical intuition of structural similarity6.
The Universe of Small Molecules of Biological Interest
The true “universe” of small molecules of biological interest is unknown, as it includes small molecules yet to be discovered7. To approximate this space, the researchers used a combination of 14 molecular structure databases that are frequently used in (untargeted) metabolomics, environmental research, and natural products research. From this extensive set of more than 700,000 biomolecular structures they uniformly subsampled 20,000 structures to represent the data space of interest.
Checking Training Datasets For Coverage Bias
With the help of UMAP embeddings and MCES distances, they tackled the question of whether datasets used for machine learning are uniform subsamples of the biomolecular space. They examined ten public molecular structure datasets frequently employed in training and evaluating machine learning models (see box). SIRIUS was trained on (an even extended version of) the MS/MS dataset, which includes molecular structures from GNPS, MassBank, and NIST.
One must exercise significant caution when drawing conclusions on the structure of the data based solely on a two-dimensional UMAP embedding8. However, if non-uniformity is apparent in the two-dimensional embedding, it is reasonable to infer that the dataset is likely not a uniform subsample in higher dimensions either. The analysis revealed that the datasets often fail to provide a representative subset of biomolecular structures, with significant portions of the biomolecular structure space entirely absent. Arguably the best coverage in the two-dimensional space was observed for the toxicity datasets and the MS/MS dataset, although this does not automatically imply that these datasets contain a uniform or representative subset of molecular structures in the higher dimensional space. Conversely, UMAP plots can be considered a useful diagnostic tool to identify datasets that are not representative of the broader biomolecular structure space.
BACE: 1513 synthetic human BACE-1 inhibitors
BBBP: 2039 molecules curated from the scientific literature discussing blood-brain barrier penetration
ClinTox: 1478 molecular structures comparing FDA-approved drugs from SWEETLEAD database with drugs that failed clinical trials for toxicity reasons retrieved
Delaney: 1128 compounds with water solubility data
Lipo: lipophilicity data for 4200 compounds (part of the ChEMBL database)
SIDER: 1427 drugs with side effects grouped in 27 system organ classes
SMRT: 80,038 small molecules and their experimentally acquired reverse-phase chromatography retention times
ToxCast: extended dataset from the Tox21 program that includes toxicology data based on in vitro high-throughput screening of 8576 compounds
Tox21: samples from 12 toxicological experiments of in vitro bioassays and a total of 7831 molecular structures
MS/MS: 18,848 unique molecular structures from GNPS, MassBank, and NIST 17
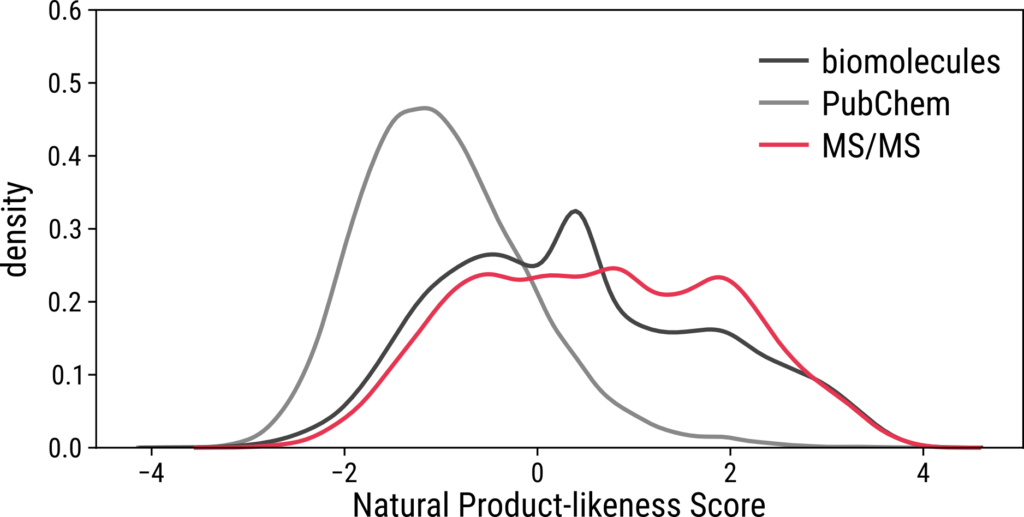
Natural Product-Likeness Of The Training Set
Analyzing natural product-likeness score distributions provides further insights into the representativeness of a dataset. This score measures how structurally similar molecules are to natural products9, enabling differentiation between biomolecules and synthetic molecules. While highly interesting molecules may have high natural product-likeness scores, their inclusion in datasets is often limited by factors such as the difficulty and cost of chemical synthesis. If the natural product-likeness score distribution of a dataset aligns with that of the biomolecular structures, it suggests that the dataset includes sufficient numbers of biologically relevant molecules. However, if the distribution resembles or exceeds that of PubChem10, which contains biomolecular structures alongside millions of synthetic compounds, the dataset likely includes only a small fraction of biomolecules. In three of the evaluated datasets—BACE, Lipo, and SMRT—the natural product-likeness score distributions differ substantially from those of biomolecules, indicating that these datasets contain molecular structures of limited biological interest.
High-Quality Training Data Is The Foundation Of Successful Machine Learning
Many widely used datasets for small molecule machine learning fail to evenly represent the diversity of biomolecular structures. As a result, machine learning models trained on these datasets may struggle to generalize effectively, as they are not exposed to the full spectrum of molecular types they might encounter in real-world applications. We strongly recommend evaluating the quality of the training dataset to identify biased molecular structure distributions, which could pose risks to model performance. This step can help assess whether models are genuinely making reliable predictions or merely adapting to idiosyncrasies in the data. Without such scrutiny, performance gains from complex machine learning models may reflect overfitting to the dataset rather than improvements that translate to practical applications. However, the absence of detectable peculiarities in a dataset does not guarantee that it is free from representational biases.
Fleming Kretschmer, Jan Seipp, Marcus Ludwig, Gunnar W. Klau & Sebastian Böcker.
Coverage bias in small molecule machine learning.
Nat Commun. 2025 Jan 9;16(1):554. doi: 10.1038/s41467-024-55462-w. PMID: 39788952; PMCID: PMC11718084.
References
- Ekins, S., Puhl, A.C., Zorn, K.M. et al. Exploiting machine learning for end-to-end drug discovery and development. Nat. Mater. 18, 435–441 (2019). https://doi.org/10.1038/s41563-019-0338-z ↩︎
- McInnes, L., & Healy, J. (2018). UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. ArXiv e-prints. https://doi.org/10.48550/arXiv.1802.03426 ↩︎
- Kretschmer, F., Seipp, J., Ludwig, M. et al. Coverage bias in small molecule machine learning. Nat Commun 16, 554 (2025). https://doi.org/10.1038/s41467-024-55462-w ↩︎
- Tanimoto, T. T. (1958). An elementary mathematical theory of classification and prediction by T.T. Tanimoto. New York : International Business Machines Corporation ↩︎
- Flower, D.R. (1998). On the Properties of Bit String-Based Measures of Chemical Similarity. J. Chem. Inf. Comput. Sci., 38, 379-386. ↩︎
- Ehrlich, H. and Rarey, M. (2011). Maximum common subgraph isomorphism algorithms and their applications in molecular science: a review. WIREs Computational Molecular Science, 1(1), 68-79. https://doi.org/10.1002/wcms.5 ↩︎
- Dobson, C. Chemical space and biology. Nature 432, 824–828 (2004). https://doi.org/10.1038/nature03192 ↩︎
- Chari T, Pachter L (2023) The specious art of single-cell genomics. PLoS Comput Biol 19(8): e1011288. https://doi.org/10.1371/journal.pcbi.1011288 ↩︎
- Ertl P, Roggo S, Schuffenhauer A. Natural product-likeness score and its application for prioritization of compound libraries. J Chem Inf Model. 2008 Jan;48(1):68-74. https://doi.org/10.1021/ci700286x ↩︎
- Kim S, Thiessen PA, Bolton EE, et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016 Jan 4;44(D1):D1202-13. https://doi.org/10.1093/nar/gkv951 ↩︎



